Наследственные болезни - это структурные или функциональные патологические изменения организма, вызванные изменением наследственной информации. В настоящее время известно более двух тысяч наследственных болезней. Согласно одной из классификаций, их можно разделить на генные, хромосомные и мультифакториальные (болезни с наследственным предрасположением).
I. Генные болезни (молекулярные болезни)
Генные болезни являются результатом генных мутаций: изменение структуры молекулы ДНК изменение РНК изменение полипептидной цепи изменение белка (структурного или белка фермента). Мутации возникают в аутосомах и половых хромосомах, они могут быть доминантными и рецессивными.
По доминантному типу наследуются болезни, связанные с нарушением структурных, транспортных белков, белков-рецепторов и др. - это болезни костной системы и соединительной ткани (ахондроплазия, синдром Марфана, нейрофиброматоз и др.). Для диагностики этих болезней иногда используют и данные электронной микроскопии (изменение мышечных волокон, клеток эпидермиса, хондроцитов).
По рецессивному типу (аутосомному или сцепленному с полом) наследуются многие болезни обмена веществ - энзимопатии или ферментопатии. Они возникают в результате мутации генов, ответственных за синтез ферментов (Д. Бидл, Э.Тейтум). У человека большинство ферментопатий вызвано мутациями структурных генов, что приводит к качественному, а не количественному изменению ферментов. Наиболее распространены энзимопатии, обусловленные нарушением аминокислотного обмена (фенилкетонурия), жирового (амавротическая идиотия), углеводного (галактоземия, фруктозурия ), пуринового и пиримидинового (синдром Леш-Нихана), минерального (нарушение обмена меди - болезнь Вильсона-Коновалова).
Моделью для изучения молекулярных болезней может служить фенилкетонурия: известна локализация гена в 12 хромосоме (определяющего синтез фермента фенилаланингидроксилазы); тип наследования (аутосомно-рецессивный); частота распространения мутантного гена в популяциях; метаболизм фенилаланина у здоровых (превращение фенилаланина в тирозин под влиянием фермента фенилаланингидроксилазы) и больных людей (некоторая часть фенилаланина из-за изменения фермента превращается в токсическую фенилпировиноградную кислоту); разработана диагностика (выявление фенилаланина в крови и фенилпировиноградной кислоты в моче); лечение (специальная диета и использование гидролизата белка без фенилаланина); профилактика (предупреждение браков между гетерозиготами); описаны четкие фенотипические проявления мутантного гена у гомозигот (выраженная умственная отсталость, изменение пигментации волос и кожи - голубоглазые блондины со светлой кожей).
Для многих генных болезней (например, серповидно-клеточной анемии) известен механизм возникновения на уровне гена, белка, фенотипа. Мутация структурного гена приводит к изменению полипептидной цепи глобина, замене глутаминовой кислоты на валин. В условиях гипоксии полипептидные цепи глобина полимеризуются, образуя длинные тяжи и эритроциты принимают форму серпа. Изменение их формы приводит к изменению функции.
Для диагностики генных болезней (энзимопатий) применяют биохимический метод и метод амниоцентеза.
II. Хромосомные болезни
Хромосомные болезни возникают при нарушении кариотипа (нормальный кариотип человека 46,ХХ, 46,ХУ), которое может быть вызвано изменением числа (геномные мутации) или структуры (хромосомные мутации) аутосом и половых хромосом.
При хромосомных болезнях имеется определенный комплекс стабильных аномальных признаков - симптомов, который входит в понятие синдром. Синдромы характеризуются определенной частотой проявления, продолжительностью жизни детей и средним весом при рождении, внешними морфологическими признаками, пороками развития внутренних органов, функциональными симптомами, дерматоглификой и определенным кариотипом.
Среди хромосомных болезней, связанных с изменением числа половых хромосом (моносомии, полисомии), наиболее часто встречаются синдромы Клайнфельтера (47,ХУ), Шерешевского-Тернера (45,Х), трисомия по Х-хромосоме (47,ХХХ), полисомия по У-хромосоме (47,ХУУ); может наблюдаться полисомия по Х-хромосоме и У-хромосоме одновременно ( 48,ХХУУ).
Эффект мутаций аутосом различен: при геномных мутациях 1-12 хромосом возникают аномалии, несовместимые с жизнью; 13-18 - полулетальные мутации (спонтанные аборты, множественные уродства, незначительная продолжительность жизни родившихся от нескольких недель до нескольких лет - синдром Патау (47,ХХ/ХУ + 13), синдром Эдвардса (47,ХУ/ХХ + 18). Трисомия по 21 хромосоме - синдром Дауна (47,ХХ/ХУ + 21) - является аномалией, совместимой с жизнь.
При анализе синдромов по половым хромосомам и аутосомам показано, что при аномалиях по половым хромосомам сохраняется нормальный интеллект или отмечается его снижение, но в большей степени нарушается развитие половых органов и гормонозависимый рост (выше или ниже средней нормы). Моносомия Х встречается реже (1: 25ОО), чем полисомии ХХУ (1: 700) и ХХХ (1: 1000).
Хромосомные аберрации в основном представлены делециями и транслокациями: при делеции короткого плеча 5 хромосомы (46,ХХ/ХУ 5р-) наблюдается синдром «кошачьего крика» (название обусловлено сходством плача ребенка с мяуканьем кошки), происхождение которого вызвано нарушением центральной нервной системы, а не аномалией голосового аппарата. Встречаются делеции по 13, 18, 21, 22 хромосомам (46,ХХ/ХУ 13q- - синдром Орбели). Транслокация 15/21 хромосом приводит к возникновению синдрома Дауна; а 9/22- хроническому миелолейкозу.
Для диагностики хромосомных болезней используется цитогенетический метод (кариотипирование, определение полового хроматина); методы амниоцентеза и дерматоглифики.
Хромосомные болезни не наследуются, так как у больных нарушена репродуктивная функция, но синдромы появляются в каждом поколении с определенной частотой как результат вновь возникших мутаций у здоровых людей.
III. Мультифакториальные болезни
Развитие некоторых болезней (гипертоническая болезнь, атеросклероз, язвенная болезнь, определенные формы диабета, шизофрения и др.) зависит как от генотипа (полигенное наследование), так и от внешней среды. Эти болезни называются болезнями с наследственным предрасположением. Для них характерно изменение нормы реакции на действие факторов внешней среды (при предрасположенности к диабету изменяется норма реакции на крахмал и сахар - увеличение глюкозы в крови).
Для мультифакториальных болезней характерно:
- отсутствие типичного расщепления, как при моногенном наследовании;
- различие в частоте заболеваний у родственников первой и второй степени родства;
-широкое варьирование количественных признаков (размах изменчивости) среди населения;
- зависимость проявления заболеваний от возраста, пола, питания и др.;
- отсутствие резких различий в проявлении признаков между здоровыми и больными людьми.
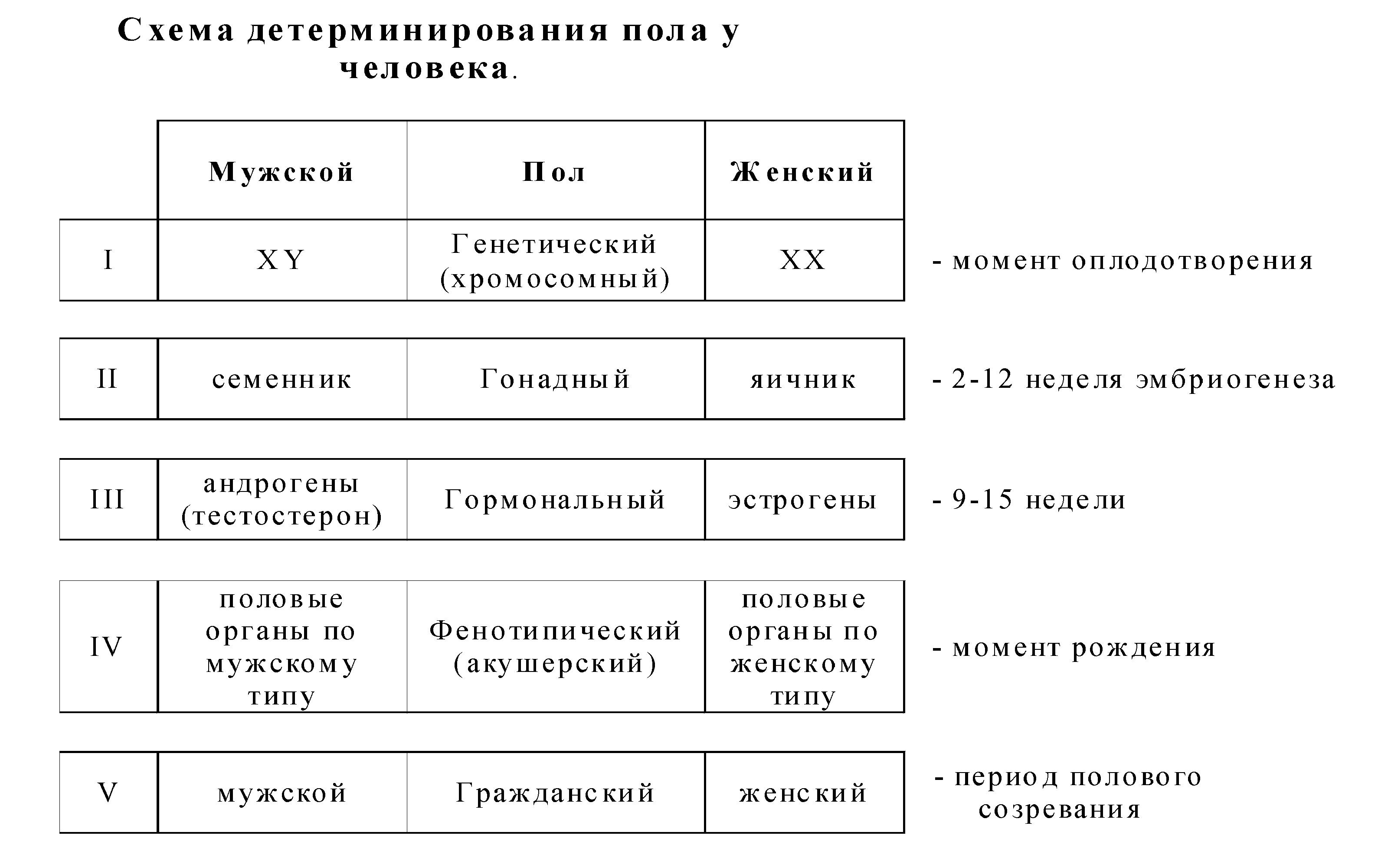
IV. Определение и развитие пола
Пол - это совокупность морфологических, физиологических, биохимичеких, поведенческих и других признаков организма, обусловливающих репродукцию.
Определение пола происходит в момент оплодотворения и зависит от набора половых хромосом в зиготе (46,ХХ/46,ХУ). В дальнейшем наблюдается дифференцировка пола: образование гонад (семенников и яичников) - формируется гонадный пол. Гонады вырабатывают гормоны, в клетках-мишенях происходит взаимодействие молекул гормона с белками рецепторами - формируется гормональный пол, что обеспечивает в последующем формирование вторичных половых признаков по мужскому или женскому типу (акушерский или фенотипический пол).
В дифференцировке мужского пола участвует андроген (находится в У-хромосоме), который детерминирует образование НУ- антигена, а он, в свою очередь, определяет развитие семенников. Наряду с андрогеном, в этом участвут ген tfm, который определяет образование в цитоплазме клеток-мишеней белков-рецепторов, взаимодействующих с тестостероном. При мутации гена tfm наблюдается синдром тестикулярной феминизации (синдром Морриса) - женский фенотип при мужском кариотипе.
Для нормального развития пола необходимо наличие двух половых хромосом (ХХ или ХУ). При геномных мутациях (хромосомные болезни) наблюдается их увеличение или уменьшение, что приводит к недоразвитию первичных и вторичных половых признаков.
V. Профилактика возникновения наследственных болезней
Профилактика возникновения наследственных болезней проводится по нескольким направлениям:
- на уровне популяций - уменьшение действия мутагенных факторов, применение антимутагенов, разработка и использование специальных скрининг-программ на наследственные болезни (диагностика и профилактика болезней обмена, хромосомных болезней);
- на уровне семей (медико-генетическое консультирование).
Медико-генетическое консультирование складывается из нескольких этапов:
I. Диагностика заболевания:
- установление природы наследственного заболевания (генное, хромосомное);
- обследование больного с применением соответствующих методов изучения генетики человека (генеалогического, цитогенетического, биохимического и др.);
- выяснение уровня нарушения наследственного материала: на уровне молекулы ДНК, хромосом, генома;
- при генных болезнях - установление связи между патологией и изменением белка (структурного или белка-фермента);
- при хромосомных болезнях - установление связи между патологией и типом хромосом (аутосомы или половые хромосомы);
- сопоставление полученных данных у обследуемого больного с фенотипической картиной известных наследственных болезней.
II. Определение вероятности развития болезни в потомстве при моногенном (аутосомно-доминантное, аутосомно-рецессивное и сцепленное с полом) и полигенном типе наследования.
III. Заключение: рекомендации врача-генетика родителям (сопоставление риска рождения ребенка с тяжестью заболевания и возможными социальными последствиями). Определение генетического риска складывается из 3 моментов: вероятность появления данной аномалии (в процентах от 1 до 100 при аутосомно-доминантном и аутосомно-рецессивном, сцепленном с полом наследовании); оценка тяжести медицинских и социальных последствий; перспектива применения и эффективность методов пренатальной диагностики.
Медико-генетическое консультирование может проводиться при обследовании вступающих в брак, обследование матери во время беременности (пренатальная диагностика эмбриона или плода), обследование ребенка после рождения.